I’m currently working through the preprocessing pipeline for the datasets. Some questions in this regards. Thank you in advance!
I read Pramod’s helpful post on the OLINK data, which was valuable to understand how to think about the LOD. Still, I have three different values for each protein (npx_protein_expression, npx_limit_of_detection, and npx_quality_control), and I’m not sure the best way to incorporate all three. Right now, I’m thinking: -remove all proteins where npx_quality_control=’warning’, -keep all values for genes with expression>LOD as is, and -do something with the values where expression <= LOD. Not sure what that something is as I don’t feel completely qualified to choose between the options that Pramod lists. Any suggestions to start?
Confirming for the missing cell types in a given sample in the CyTOF data - these are truly missing, and not ‘0’ or undetectable?
I have a follow-up (similar) question for the antibody data - there is a field ‘lower_limit_of_detection’ that is often greater than the value in the field ‘ab_titer’. How is it recommended to handle these instances?
Hi! Sorry for the late reply, not sure what Jeremy Gigi tried to do but I simply used the npx_protein_expression as is. What you suggest seems like a good idea especially since the paper from which this data derived excluded protein levels below the LOD. I’ll have to incorporate this into my own pipeline. Here’s the description they give in the paper about processing the data:
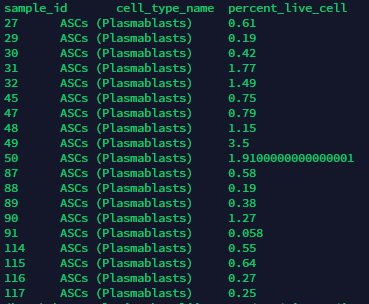
As for missing cell types I’m not sure what you’re referring to, if you dump the table from the API you should get something like this:
At some point I transform the table into subject_id versus cell type specific percent_live_cell but I don’t recall having missing data. Did you try something like this and then you found some missing percent_live_cell values?
As for the Ab titers, I also used these values as is and the paper doesn’t seem to suggest much use of this lower limit. Not sure if the idea was to let people choose whether to perform this filtering or not.
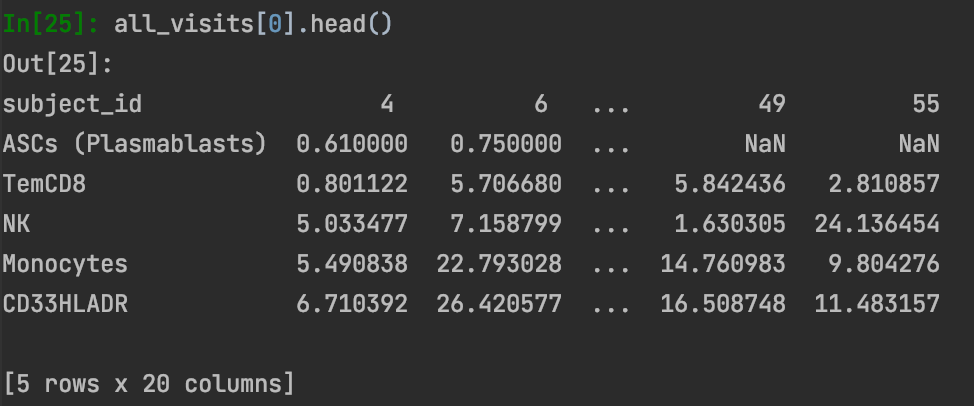
Indeed, when I did the transformation from edgetable to matrix (I made a separate cell type x subject matrix for visits 1-5), I found NAs. This is from visit 1:
Thank you for posting the methods description. The current data has max 6aP and 10wP donors per visit, so I think we already have the 2 subjects they mention excluded. Even after filtering out instances where npx_quality_control!=‘Pass’, I still have 276 proteins for a given day if I exclude subjects where there is NA (this is why, per day, I may have < 16 subjects). I haven’t done additional filtering on the LOD, but I agree, would make sense to follow their protocol. Thanks!